病史摘要
患者男性17岁,因“高血压2年,高血钾1周”入院。根据典型临床表型高血钾、高血压、高血氯、代谢性酸中毒,同时肾小球滤过率正常、肾功能正常,小剂量噻嗪类利尿剂治疗有效,且基因检测示WNK4(P561L)突变,诊断为假性醛固酮减少症Ⅱ型(PHA Ⅱ)散发病例,其相关类型在国内外尚属首例。治疗为终身服用小剂量噻嗪类利尿剂。该病历提示,重视高血钾、高血压的鉴别诊断,注意肾功能和血容量改变;该病为常染色体显性遗传,须行家系调查、明确遗传方式、探查基因缺陷;早期诊断、合理治疗、早期纠正水电解质异常可获良好预后。
病历简介
患者,男,17岁,因“发现血压高2年,血钾高1周”入院。患者2年前在家中偶测血压,发现血压高(140/90 mmHg),当时未引起重视,未就医,后多次测血压波动于150~140/100~80 mmHg。1年前患者就诊于当地医院,先后服用美托洛尔、硝苯地平、赖诺普利治疗,但血压仍不能控制到理想水平,3个月前自行停药。
病程中患者无阵发性头痛、头晕、心悸,无消瘦、怕热、多汗。1周前在外院查血钾6.7 mmol/L,无发作性四肢无力、抽搐;查心电图无异常,血肌酐正常,未治疗。患者足月顺产,生长发育、智力与同龄人相比无异常。父母非近亲婚配,家族中无类似病例。
体格检查
BP 145/95 mmHg,体质指数(BMI)25.14 kg/m2 ,神清,发育正常,体型均匀性肥胖,全身皮肤黏膜无黄染及出血点,无满月脸。两肺呼吸音清,心率80次/分,律齐,各瓣膜区未及病理性杂音。腹平软,无压痛,肝脾肋下未及,未闻及血管杂音。脊柱四肢无畸形及活动障碍。外生殖器发育正常。生理反射存在,病理反射未引出。
实验室及辅助检查
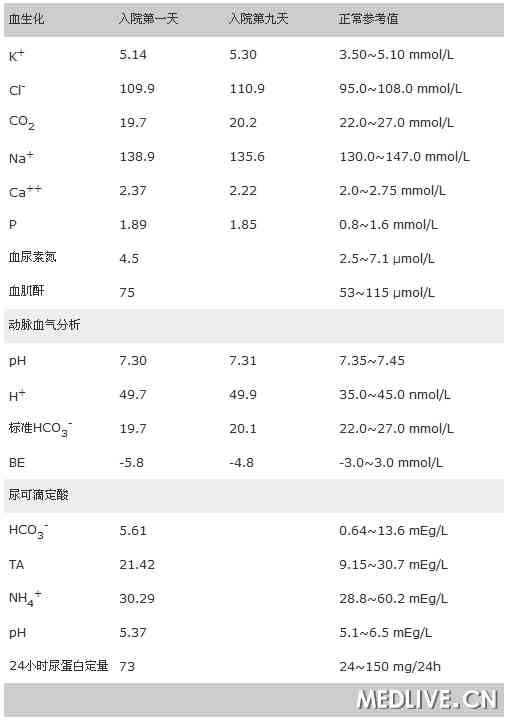
血尿粪常规正常。入院后测定患者相关生化指标,并对部分项目进行复查,结果见表1。肾功能:血肌酐、尿素氮、尿微量白蛋白正常,尿红细胞阴性。
B超:双肾大小形态正常,输尿管、膀胱未见异常。同位素肾图、肾血流图正常。锝(99m TC)DTPA 肾动态显像:肾小球滤过率(GFR) 左肾47.68 ml/min,右肾51.27 ml/min,提示双肾GFR正常。
尿可滴定酸、同步血气分析结果见表1。
血浆肾素活性、血醛固酮正常。血皮质醇昼夜节律:8 Am 15.50 μg/dl (正常值7~22 μg/dl),4 Pm 5.20 μg/dl,0 Am 2.3 μg/dl。24小时尿皮质醇156.2 μg/24h尿。血变肾上腺素、血去甲变肾上腺素正常;尿游离肾上腺素、尿游离去甲肾上腺素、尿游离多巴胺均正常。
肾上腺CT:双侧肾上腺未见异常。心电图正常。
表1 血尿生化检查结果
诊治经过
诊断 假性醛固酮减少症Ⅱ型(PHA Ⅱ) 。
综合分析患者的临床特征,高血压、高血钾、高血氯、代谢性酸中毒,且不存在肾功能障碍,与假性醛固酮减少症Ⅱ型(PHA Ⅱ)的临床特征相符,临床诊断基本明确。
考虑PHAⅡ是常染色体显性遗传病,在获得知情同意后,我们进行了家系调查,对患者及其亲属的外周血DNA进行丝氨酸/苏氨酸蛋白激酶基因WNK1和WNK4的突变检测。
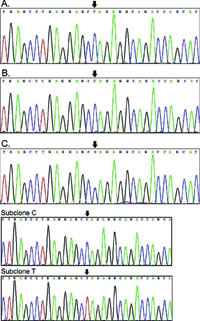
结果该患者WNK4基因第7号外显子存在错义突变Pro561Leu(见图1)。WNK1基因的印迹杂交结果显示,患者WNK1基因1号内含子未显示杂交差异。而患者的父母、祖父母、外祖父母都不存在慢性高钾血症,且不存在WNK4相同类型的基因突变。另外,100名非亲缘正常人外周血DNA样本中均未发现上述突变。
该17岁男性患者具有典型的临床表型,并且结合基因检测结果证实为一PHA Ⅱ散发病例,其相关突变类型WNK4(P561L)在国内外尚属首例。
给予患者氢氯噻嗪(双氢克尿塞)25 mg,每日2次口服,1周后患者的高血压和代谢异常(高血钾、高血氯、代谢性酸中毒)均被完全纠正。建议患者在定期随访前提下,终身服药治疗。
讨论
假性醛固酮减少症Ⅱ型(PHAⅡ),又称家族性高血钾高血压(FHH),是一种常染色体显性遗传病。患者的主要临床特征包括高血钾、高血压、高血氯、代谢性酸中毒,同时肾小球滤过率正常。另外小剂量噻嗪类利尿剂治疗即可很好地纠正相关的代谢异常。
PHA II发病极为罕见,确切的发病率尚不清楚,1964年佩弗(Paver)首次报告了一个散发病例,到目前为止,文献中报告过的家系仅仅约20个,散发病例10余个。
PHA II的相关变异基因
1997 年曼斯菲尔德(Mansfield)等对PHA Ⅱ发病的遗传学机制进行研究,分析8个家系,明确了两个相关基因座位。之后,迪塞(Disse-Nicodeme)等又测定出第三个,由此将PHAⅡ分为三型,PHA ⅡA(Chr.1q31-q42)、PHA ⅡB(Chr.17p11-q21)和 PHA ⅡC(Chr.12p13)。2001年威尔森(Wilson)等人经定位克隆证实后提出,PHAⅡ的发病与WNK1和WNK4基因的突变有关。
图1 WNK4基因7号外显子直接及亚克隆测序结果
A为患者母亲,B为患者父亲,C为患者。箭头所指为7号外显子561位密码子第2个碱基,在患者母亲(A)和患者父亲(B)为核苷酸C,而患者本人(C)为杂合突变C>T,这一突变使脯氨酸转变为亮氨酸。亚克隆C和亚克隆 T为亚克隆处理后的C>T突变。
基因突变的类型
迄今明确基因突变类型的家系仅七个,各自具有不同的突变类型。WNK1基因定位于Chr.12p13.3 ,WNK1的突变是一号内含子内大片段碱基的缺失,在两个PHAⅡ家系中分别发现WNK1基因一号内含子内22 kb和41 kb大片段的缺失,前者的缺失包含在后者的序列中。 WNK4基因定位于Chr.17p11-q21, 在5个家系中分别明确不同的突变类型,其中四种(Glu 562 Lys,Asp 564 His, Asp 564 Ala,Gln 565 Glu)和我们新发现的一种Pro561Leu都位于一个包含10个氨基酸的WNKs蛋白激酶的酸性保守序列(EPEEPEADQH)中,这个序列位于WNKs蛋白激酶第一个螺旋区的远端;另一种突变(Arg1185 Cys)位于WNKs蛋白激酶第二个螺旋区的远端。
基因突变导致临床症状
后来的许多相关研究试图揭开其发病机制,结果表明WNK1和WNK4基因的突变可导致WNK1或WNK4活性改变,进而引起肾小管上皮多种转运蛋白功能紊乱,引起Na+、Cl-的重吸收增加,泌K+减少,进而导致了PHA Ⅱ的一系列主要表现,包括高血钾、高血氯、水钠潴留、血容量扩张,前两项又导致代谢性酸中毒。
由于PHAⅡ的肾小管对Na+、Cl-的重吸收增加,泌K+障碍,患者存在不同程度慢性高血钾、高血氯和容量依赖性高血压。长期处于高血钾状态,患者可以无明显的主诉症状、阳性体征和心电活动的异常。有的患者以肌肉痛性痉挛和高血钾性周期麻痹起病。
与高血钾不同,高血压易被察觉,是患者求诊的主要原因,患者高血压发生的时间有较大的个体差异。在已报道的家系中,所有患者都有明显的高血钾,而大多数在青少年时期没有血压异常,该现象可能和机体的代偿机制及环境因素有关,随着年龄的增长,血管的弹性变差,代偿能力下降,发生高血压并且逐渐加重。
在内分泌激素检查方面,因为患者是容量依赖性高血压,表现为低或正常肾素活性,而醛固酮水平一般正常或略高于正常,原因可能是高血钾促使醛固酮分泌增多。
PHA II的临床表现常不具特征性
PHA Ⅱ的临床表现有上述特殊性,诊断时,必须具有高血钾,再结合高血氯、代谢性酸中毒、高血压,同时肾功能正常,临床上基本能够明确诊断。由于这些临床表现分开并不具有特征性,很多疾病可以出现一部分症状体征,因此需要排除其他一些疾病,如慢性肾功能衰竭、Ⅳ型肾小管酸中毒、原发性高血压、药物影响等,临床鉴别主要是观察肾功能和血容量改变。
本病的病因是遗传基因异常,呈常染色体显性遗传,对疑似患者必须进行家系调查,分析临床资料,明确其遗传方式,探查基因缺陷,实现在分子水平上的诊断,这些工作不仅可以进一步明确诊断,还为患者生育健康后代提供选择的机会。
PHA II的病情易被掩盖
PHA Ⅱ的发病率可能比目前所知要高一些。有些患者PHA Ⅱ的临床表现不典型,血钾没有超过正常上限,或有可能是目前血钾的上限不一定恰当,使一部分患者偏高的数值被认为是正常而忽视;另一方面,噻嗪类利尿剂在原发性高血压中应用比较广泛,从而使PHAⅡ的病情被掩盖,增加了诊断PHAⅡ的难度,这些问题可能都会导致PHAII临床诊断率低。结合以上研究结果,在诊断原发性高血压和处方治疗前,特别是针对年轻的高血压患者,有必要进行血电解质的筛查。
早期诊断合理治疗愈后良好
对PHA Ⅱ认识的初期,虽然病因和发病机制不清,由于患者有高血压和高血钾而给予小剂量噻嗪类利尿剂治疗,结果发现患者的代谢异常(包括高血钾、高血氯、高尿钙、代谢性酸中毒)被很好的纠正,血压也恢复到正常水平。有研究者指出,噻嗪类利尿剂(如氢氯噻嗪25~100 mg qd),经过约一周的作用时间即可达到上述疗效。
由于存在遗传缺陷,PAH Ⅱ患者需要终身服用小剂量噻嗪类利尿剂。 PHA Ⅱ患者若早期得到诊断和合理治疗,其水电解质代谢异常早期就能纠正,预后良好。
小结
上文分析了PHAⅡ这一罕见的单基因病的分子基础,并讨论了该病所致肾脏调节水盐代谢异常的机制,具有非常重要的意义,为治疗原发性高血压药物的开发提供新的靶点。
目前发现的PHA Ⅱ的家系较少,在已知家系中,还有一些家系的致病基因尚未明确;不同突变造成的表现型存在细微差别,表现型和基因型之间的关系尚不清楚;WNK4突变主要集中在一个酸性结构域,该区域主要发挥怎样的功能,如何发挥功能至今仍不清楚,这些都值得进一步研究。
在临床工作中,应重视高血钾、高血压的鉴别诊断,需要提高对PHA Ⅱ的认识,以早期诊断和治疗,对患者的预后和优生优育有重要意义。
(作者:上海瑞金医院内分泌代谢科 龚慧 汤正义 孙立昊 宁光)

![京卫网审[2010]第0050号](http://999120.net/cert/wwsz20110722.gif){kind=link}